

Hiperplasia supra renal congénita
Introdução
Definição
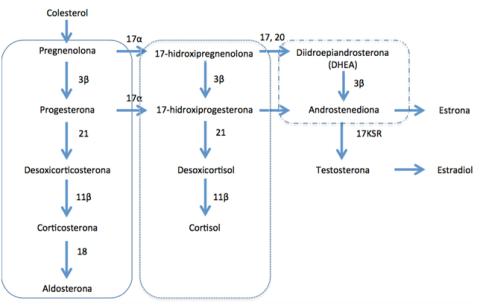
A Hiperplasia Suprarrenal Congénita (HSRC) é uma patologia autossómica recessiva, que se traduz em défices enzimáticos específicos na síntese do cortisol e/ou aldosterona (esteroidogénese supra-renal – ver Figura 1).(1)
Epidemiologia
A deficiência de 21-hidroxilase, resultante de mutações no gene CYP21A, é a forma mais frequente de HSRC, responsável por 90-95% dos casos. A incidência da forma clássica é de 1:10000 a 1:20000 recém-nascidos, contudo a forma não clássica é mais frequente apresentando incidência aproximada de 1:1000, ambas com variações étnicas e raciais significativas. (1-3)
Manifestações Clínicas
O fenótipo clínico depende do sexo, da localização do bloqueio na síntese, da gravidade das mutação genéticas e da atividade enzimática residual.
Formas clássicas perdedoras de sal
Cerca de 75% dos doentes com HSRC devido a deficiência da 21-hidroxilase têm défice de aldosterona para além do défice de cortisol e por esse motivo a crise adrenal aguda pode ser uma forma de apresentação. Nesses casos, nas primeiras 4 semanas de vida (geralmente entre o 7-14º dias), pode surgir quadro de má evolução ponderal, hiporreatividade, dificuldades alimentares, vómitos, desidratação, hipotensão, hipotermia, e hiperpigmentação. Analiticamente são característicos a hiponatremia, hipercaliemia, hipoglicemia e acidose metabólica. Esta situação pode evoluir para choque e ser fatal. Nos casos em que o hipoaldosteronismo é mais ligeiro, as únicas manifestações podem ser a má progressão ponderal e as dificuldades alimentares com ou sem hiporreatividade.(3-4)
Formas clássicas virilizantes simples
A HSRC no recém-nascido (RN) do sexo feminino pode manifestar-se por anomalias genitais externas com diferentes graus de virilização - desde clitoromegalia (clítoris do RN > 9 mm) até aspeto de phallus; e devem ser c9lassificadas segundo a Classificação de Prader (ver Quadro 1)(2). Os órgãos internos não são afetados.(2-4)
| 1 – Genitais femininos normais com clitoromegalia isolada |
| 2- Fusão labial parcial e clitoromegalia |
| 3- Fusão labioescrotal de forma que existe apenas um orifício a partir do seio urogenital e clitoromegalia |
| 4 – Fusão das pregas labioescrotais de forma que o único orifício se encontra na base da estrutura fálica |
| 5 – Virilização masculina completa com falo do tamanho de um pénis, fusão labial completa e meato urinário na glande |
No RN do sexo masculino, os genitais não estão afetados, pelo que na ausência de crise adrenal, o diagnóstico é habitualmente mais tardio. Nessas situações são detetados sinais de hiperandrogenismo precoce (2-4 anos) nomeadamente pubarca, acne e aumento do pénis com testículos normais para a idade. Observa-se também, aceleração da velocidade de crescimento (VC) e da maturação óssea.(3-4)
Formas não clássicas (tardias)
As formas não clássicas são habitualmente diagnosticadas mais tardiamente – crianças em idade escolar, adolescência e mesmo idade adulta. Manifestam-se por sinais de hiperandrogenismo como pubarca precoce, aceleração do crescimento, acne resistente à terapêutica, oligomenorreia, hirsutismo e/ou infertilidade.(3-4)
Diagnóstico Diferencial
Estenose Hipertrófica do Piloro; Disgenesia gonadal, Síndroma de Insensibilidade aos Androgéneos, Hemorragia Suprarrenal; Hipoplasia suprarrenal, Adrenarca Precoce, Doença de Addison.
Rastreio Neonatal
Em alguns países, o rastreio da deficiência de 21-hidroxilase está inserido no programa de rastreio neonatal precoce através do doseamento de 17OHP em sangue recolhido em papel de filtro (cartão de Guthrie).(2-5)
De referir que resultados falsos positivos podem ser observados em prematuros e RN doentes, pelo que é sempre recomendada a comparação com valores de referência para idade gestacional; resultados falsos negativos têm sido mais frequentes em RN do sexo feminino e em situações pós corticoterapia durante a gestação.(3-4) Um resultado positivo no rastreio neonatal deve ser confirmado por nova colheita para doseamento sérico de 17HOP e avaliação eletrolítica.
Exames Complementares
O diagnóstico de HSRC depende da demonstração de produção inadequada de cortisol e/ou aldosterona, associada a acumulação de hormonas precursoras. Idealmente, a investigação de situações suspeita de HCSR deve ser realizada a nível hospitalar, sob orientação de Endocrinologia Pediátrica.
Concentração sérica muito elevada de 17OHP numa colheita da manhã ( 3500 ng/dL e praticamente todos apresentam >1200 ng/dL). Em caso de suspeita clínica, com valores de 17OHP basais normais, a prova da ACTH é considerada o exame de eleição para o diagnóstico; contudo não é imprescindível, nem deve fazer protelar o início da terapêutica.(4)
| 17OHP basal (ng/dL) | 17OHP pós ACTH (ng/dL) |
|---|---|
| 10000 HSRC forma clássica provável |
10000 HSRC forma clássica |
| 200-10000 HSRC forma não clássica provável |
1000 -10000 HSRC forma não clássica |
| HSRC pouco provável | Não afetado ou heterozigoto |
Para melhor caracterizar o defeito enzimático e definir implicações terapêuticas, é aconselhado o doseamento de outros precursores da cascata da esteroidogénese, nomeadamente cortisol, desoxicorticosterona, 11-desoxicortisol, 17-hidroxipregnenolona, dihidroepiandrosterona e androstenediona. Devem também ser avaliados: glicemia, ionograma, estudo ácido base e Atividade da Renina Plasmática (ARP).(3-4)
Estudo genético: a correlação fenótipo-genótipo é razoável, pelo que conhecer as mutações pode ajudar a predizer a gravidade. Estão descritas múltiplas mutações causadoras de HSRC, com atingimento variável de diferentes enzimas da esteroidogénese e que levam a expressões distintas da doença.(5) Segundo as orientações atuais, o estudo de genético não deve ser realizado por rotina, exceto pela necessidade de aconselhamento genético, diagnóstico pré-natal ou quando a prova da ACTH foi inconclusiva.(2-4)
Outras avaliações:
Ecografia pélvica no RN do sexo feminino com genitália ambígua para avaliar presença de útero ou anomalias renais e cariótipo de sangue periférico (confirmação do sexo genético).(3)
Tomografia computorizada da glândula suprarrenal - pode ser útil se existir suspeita de hemorragia suprarrenal em RN com sinais de falência suprarrenal e sem sinais de virilização.
Idade óssea - na avaliação de crianças com pubarca precoce, clitoromegalia e aceleração do crescimento.
Tratamento
Neonatal
Indicado perante situações de diagnóstico pré-natal, rastreio neonatal positivo, genitália ambígua com ausência de gónadas palpáveis e nas situações de crise adrenal aguda. Deve ser iniciado logo após a realização da colheita para doseamentos hormonais, nomeadamente de 17-OHP, ionograma, glicemia e ARP.
Crise adrenal aguda perdedora de sal
É uma urgência médica. Pode ocorrer no período neonatal ou em situações de stress/doença.
Deve incluir: correção da hipotensão/desidratação, do desequilíbrio glicoeletrolítico e do défice de cortisol; deve ser endovenoso.
- Se choque - volemização inicial com bólus de soro fisiológico (20 ml/kg, repetir se necessário);
- Correção da desidratação + manutenção - NaCl 0,90% com glicose a 5-10% consoante glicemias, ao ritmo de 1,5-2x as necessidades basais;
- Correção da hipoglicemia – bólus de glicose 10% (2-4ml/kg) se hipoglicemia persistente;
- Correção da hipercaliémia – geralmente corrigida com a perfusão de glicose e pelo efeito mineralocorticóide das altas doses de glicocorticóide; se refratária ponderar gluconato de cálcio, insulina/glicose, bicarbonato de sódio, salbutamol, resina permutora de iões ou diálise.
- Glicocorticóide (GC) - bólus de hidrocortisona (HC) – dose de stress (50-100 mg/m2SC/dose; dose habitual no RN 25 mg); seguido de administração de HC na mesma dose repartida 6/6h. Após as 24-48h passar a 50 mg/m2SC/dia 8/8h.
Durante o tratamento endovenoso com altas doses de GC não é necessário administrar mineralocorticóide (MC). Quando o RN estiver estável, com tolerância alimentar adequada, deve iniciar tratamento de manutenção.
Tratamento de Manutenção
- Glicocorticóide: o GC aconselhado é a HC, quer pelo menor dano no crescimento, quer pela sua atividade mineralocorticóide. Dose 10-20 mg/m2SC/dia; ideal 10-15 mg/m2SC/dia, dividida em 3 tomas diárias; a formulação em suspensão oral não deve ser utilizada.
- Mineralocorticóide e suplementação de sódio: a suplementação com MC está indicada nas formas clássicas perdedoras de sal e também em casos de formas virilizantes simples (situações de ARP aumentada ou diminuição da Aldosterona/ARP). Contribui ainda para redução da dose de GC.
Recomenda-se a administração de fludrocortisona (Dose 0,05-0,20 mg/dia, máximo 0,4 mg/dia, dividido em 1-2 tomas). Entre os 6 e 12 meses de idade há geralmente necessidade de reduzir a dose, devido ao aumento da sensibilidade renal.
Deve também ser administrado Cloreto de Sódio (sal de cozinha) cerca de 0,5 g dividido pelas refeições, durante o 1º ano de vida (enquanto o regime alimentar não é suficiente para suprir esta necessidade).(3-4)
As situações de genitália ambígua requerem também avaliação por equipa de cirurgia especializada de forma a melhor decidir e programar intervenção caso esta esteja indicada(4).
Tratamento em situações de stress
Recomenda-se aumentar a dose de GC; não havendo necessidade de ajuste do MC.
- Em situações de doença febril (>38.5°C), gastroenterite com desidratação -> duplicar ou triplicar a dose durante a doença aguda.
- Perante intolerância oral ponderar necessidade de HC intra-muscular.
- Situações de doença aguda grave ou cirurgia major – optar por HC endovenosa (100 mg/m2SC/dia, 3-4 administrações pelo menos 24h).
Não está recomendado aumentar as doses em situações de doença aguda ligeira, stress emocional ou antes do exercício físico.(3-4)
Tratamento das formas não clássicas
O tratamento das formas não clássicas apenas está recomendado perante pubarca precoce e rápida progressão da maturação óssea com possíveis implicações na estatura final; não está recomendado iniciar terapêutica perante HCSR não clássica em indivíduos assintomáticos e deve-se descontinuar terapêutica após resolução dos sintomas.(3-4) Tem indicação para terapêutica de stress aqueles que estão sob corticoterapia e por isso com possibilidade de supressão do eixo adrenal.
Evolução
O objetivo do tratamento da HSRC é evitar o hipercortisolismo e o hiperandrogenismo, permitindo um crescimento e desenvolvimento pubertário adequados.
Como complicações possíveis desta doença e da terapêutica associada requerem especial atenção: o aumento da massa gorda, o comprometimento da estatura final, sinais de hiperandrogenismo e o risco de síndroma de Cushing iatrogénico. Apesar do efeito do GC na densidade mineral óssea ser uma preocupação, as doses administradas de HC não parecem interferir, pelo que não está recomendada a realização de densitometria por rotina. Complicações como irregularidades menstruais, infertilidade, massas adrenais ou testiculares benignas são mais frequentes nesta patologia comparativamente à população saudável pelo que implicam vigilância mais estrita.(2-4)
A melhor abordagem de forma a diminuir o impacto das complicações é a optimização terapêutica com uma monitorização rigorosa da dose mínima eficaz da corticoterapia.
Recomendações
É recomendado seguimento em consulta de endocrinologia pediátrica com monitorização mensal nos primeiros 3 meses; trimestral até aos 12 meses e depois cada 3-6 meses. A monitorização deve incluir avaliações:
- Clínica: somatometria, VC, tensão arterial, frequência cardíaca e sinais de hipercortisolismo iatrogénico.
- Analítica (idealmente de manhã antes da toma da medicação): 17 OHP, androstenediona, testosterona, ionograma e ARP. O objetivo não é normalizar a 17-OHP pois a normalização traduz hipercortisolismo, mas manter entre 500-1000 ng/dL; a androstenediona e a renina plasmática nos valores normais para a idade.
- Maturação óssea após os 2 anos anualmente, ou semestralmente se idade óssea avançada.(3)
Bibliografia
- Sperling MA. Pediatric Endocrinology. Fourth Edi. Elsevier Saunders; 2014. 471-532 p. Available from: elsevierhealth.com
- Turcu AF, Auchus RJ. Adrenal Steroidogenesis and Congenital Adrenal Hyperplasia. Endocrinol Metab Clin North Am. 2015;44(2):275–96.
- Ishii T, Anzo M, Adachi M, Onigata K, Kusuda S, Nagasaki K, et al. Guidelines for diagnosis and treatment of 21-hydroxylase deficiency (2014 revision). Clin Pediatr Endocrinol. 2015; 24(3): 77-105.
- Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2010 Sep;95(9):4133–60.
- Mendes C, Matos IV, Ribeiro L, Oliveira MJ, Cardoso H, Borges T. Hiperplasia Congénita da Suprarrenal por Deficiência de 21-Hidroxílase: Correlação Genótipo-Fenótipo. Acta Med Port. 2015;28(1):56–62.
Deseja sugerir alguma alteração para este tema?
Existe algum tema que queira ver na Pedipedia - Enciclopédia Online?